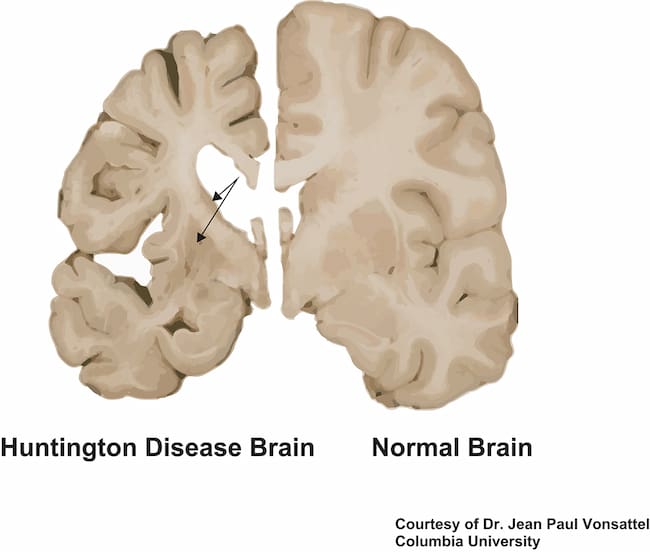
What is Huntington’s Disease
Huntington’s disease (HD) is a hereditary neurodegenerative disorder that typically produces symptoms in the 30s to 50s. There are approximately 30,000 symptomatic individuals in the United States and another 200,000 at-risk of inheriting the gene that causes HD. However, many people who carry an expanded copy of this gene never develop symptoms themselves.
The “HD gene” contains instructions for making a protein called huntingtin, which appears to be involved in communication between brain cells. In most cases, everyone has two copies of the HD gene.
In persons with Huntington’s disease, an abnormal expansion of the trinucleotide repeat sequence CAG within one of these genes results in the production of an unusually long variant form of the huntingtin protein, called an “expanded” or “mutant” form. In general, a longer polyglutamine repeat is associated with an earlier onset and more severe disease manifestations.
What are the symptoms of Huntington’s disease:
Progression of HD typically involves both motor and cognitive dysfunction. The earliest detectable deficits relate to coordination and balance (e.g., impaired performance on tandem walking, rapid alternating movements). Difficulty in speech initiation and fluency typically appears next.
Cognitive impairment becomes detectable after 5-10 years from symptom onset, starting with subtle deficits in executive function that may remain unnoticed for several years before dementia develops.
what is the first sign of Huntington’s disease:
– Excessive repeated yawning.
Huntington’s disease is a genetic disorder that results in the progressive breakdown of nerve cells in certain areas of the brain, which leads to movement abnormalities.
It is characterized by four main symptoms: muscle rigidity, uncontrolled movements, loss of intellectual faculties, and emotional disturbances. It affects more men than women and gets progressively worse over time. The cause for this condition is an expansion within chromosome 4 of the huntingtin gene, which makes the defective protein that gradually accumulates in nerve cells.
Huntington’s disease is a genetic disorder and so it runs in families. Except for the children of someone who already has Huntington’s disease, everyone else who inherits such a gene will develop Huntington’s disease at some point. The average age for someone to first notice symptoms of this condition is anywhere from thirty-five to forty-two years old.
As with many neurological disorders, men and women are affected equally by this disorder and there seems to be no significant difference between ethnic backgrounds and geographical locations where this condition appears.
Symptoms:
Not all people with Huntington’s have visible symptoms when they are diagnosed but most people experience changes in their physical and mental abilities at some point. The person may begin to have problems with walking, swallowing, remembering, and even speaking. They can become depressed or anxious and mood swings and temper outbursts may surface.
How is it inherited:
Huntington’s disease is a sporadic dominant genetic condition which means that there is no pattern regarding who inherits the defective gene. It will affect each child differently if they inherit it because of this randomness.
Sporadic means that the disorder occurs independently in people with no history of this condition in their family members while dominant refers to how one parent still has enough ‘dominant’ Huntington’s genes to pass on the disease to his/her child. Some children will thus develop symptoms while others will not, which is why it can be called sporadic.
How common is Huntington’s disease:
– Huntington’s Disease is the most common of the major hereditary disorders. It is estimated to affect 1 in 10,000 individuals.
What are the early signs of Huntington’s disease:
– The first sign of Huntington’s is often a slight tremor or fine shake in one hand. Other general symptoms can include slurred speech, clumsiness, and poor coordination. Emotional changes can also occur including depression, anxiety, and an excessive desire for things that were once enjoyed (such as alcohol).
Is there any cure for Huntington’s disease:
There is no known treatment or cure available for this condition at present although research is currently underway to find one? However, what is helpful with some patients are medications that help reduce the symptoms of the disorder.
Huntington’s disease symptoms:-
Huntington’s has a wide range of symptoms and severity can vary from patient to patient. Some people start experiencing symptoms as young as age 2, while others don’t begin showing signs until they’re in their 40s or 50s.
The disease progresses along with a specific pattern with four stages: pre-symptomatic (no signs of the disorder during this period); early symptomatic (initial symptoms such as mood swings and irritability); mid-stage (more noticeable personality changes and physical symptoms such as jerky movements; late-stage (significant loss of motor control which includes deterioration in fine motor skills)
How is Huntington’s disease treated:
There is currently no cure for Huntington’s Disease however medication may be prescribed to help manage symptoms. Some of the most commonly prescribed medications are for depression, anxiety, and nausea.
Huntington’s disease prognosis:
Many people with Huntington’s Disease can expect to live into their 50s or 60s, but some may only develop the disorder after age 60. Life expectancy will vary among individuals, depending on which gene is inherited by each child.
An individual who inherits both mutated Huntingtin genes has an earlier onset than if they only inherit one of these genes. However, individuals who carry this mutation but do not experience symptoms have also been identified so life expectancy cannot be predicted accurately at present time.
who gets Huntington’s disease:
– Huntington’s Disease (HD) is an autosomal dominant genetic abnormality that leads to degeneration of neurons in the brain. Huntington’s mutated gene (HD) is dominant because every child has a 50% chance of inheriting either mutated or non-mutated version from their parents.
The age of onset for Huntington’s ranges from 25-30 years and symptoms first appear about 10-15 years after the first signs of symptoms. Symptoms first appear before the age of 35 in approximately 75% of people with HD and before age 40 in 90%. However, some individuals may not begin showing any signs until they reach their 60s or 70s.
Individuals who carry this mutation but do not experience symptoms have also been identified so life expectancy cannot be predicted accurately at present time. Prevalence refers to how common an illness is, but not how quickly it progresses.
Genetic testing for Huntington disease:
– The only way to confirm a suspected diagnosis of Huntington’s Disease is through genetic testing to measure an abnormal protein that results from having this condition (called ‘huntingtin’).
This test requires a blood sample or cheek swab. If an individual has symptoms typical for HD there are many benefits to confirming the diagnosis as soon as possible including allowing individuals to plan for the future, helping avoid misdiagnosis, and identifying other family members who may also be at risk.
For patients in the early stage of Huntington’s disease, genetic testing is important to confirm or rule out a diagnosis. Individuals who are found not to have the mutated gene will not develop Huntington’s Disease however individuals with a confirmed diagnosis are encouraged to start planning for their future as soon as possible through life insurance, living will/trust documents, and financial planning.
Huntington’s disease vs Parkinson’s:
– Both Huntingtons Disease and Parkinson’s Disease are degenerative nerve disorders that affect the brain. However, they are two distinct diseases with different symptoms and causes.
Huntington’s is caused by an inherited faulty gene that controls cell activity in the brain whereas Parkinson’s is not inherited but rather develops from a combination of environmental factors and genetic predisposition.
Parkinsons disease affects up to 1% of people over age 65 compared to 2% of all people who have Huntington’s Disease. Individuals with both Parkinson’s Disease and Huntington’s Disease may experience more severe symptoms as these two conditions can be cumulative.
There are currently no known therapies or drugs that can delay or stop the progression of Huntington’s disease however early intervention may help reduce certain symptoms such as depression, sleep disturbances, and mood changes.
Huntington’s disease chromosome:
– The responsible gene for Huntington’s disease is located on chromosome 4. Chromosomes are rod-shaped bodies found in cells. They carry our genes and inherited information about us, physically shaped like rods or long, thin fibers wrapped around themselves. There are 46 chromosomes in each cell.
Huntington’s Disease chromosome has large CAG (cytosine-adenine-guanine) triplets repeated more than 36 times which is associated with an earlier age of onset and severity of the illness.
Huntington’s disease inheritance:
– Huntington’s disease is an autosomal dominant disorder. This means that if one of the parents has Huntington’s then there is a 50% chance that their child will develop the condition too.
There is no single inherited gene responsible for HD in families in which more than one person has been affected, however, it remains unclear how many different genes may increase susceptibility to developing this disease. The mutation results in an abnormal protein (huntingtin) which builds up and causes damage in brain cells.